Dr Björn M. Burmann describes how his group at the University of Gothenburg are elucidating how altered protein dynamics contribute to the functional repertoire.
Research in the Burmann Lab is focused on understanding how proteins in cells do their job and which biophysical rules govern their dynamics and structural adaptions; in essence how they function en detail. Just as similar rules also apply in the micro – or nanoworld, as in the macroscopic world around us, the theme ‘life is movement, and movement is life’ is of crucial importance to understanding how proteins fulfill their role.
Currently, however, a variety of approaches, such as X-ray crystallography, cryo-electron microscopy (cryo-EM), and nuclear magnetic resonance (NMR), are providing unique insights into the three-dimensional structure of proteins, and their use in the development of a more comprehensive understanding and detailed characterisation of their dynamics, such as flexibility and/or mobility of certain regions, are essential.

In the Burmann lab, we are making use of the unique capabilities of NMR spectroscopy to study the dynamics of the protein backbone as well as the amino acid sidechains, which is enabling us to get a deep view of the allosteric processes regulating the functionality of proteins. NMR’s unique ability to study a variety of states in an aqueous buffer solution provides direct access to a large variety of important biological processes which are spread over a diverse set of timescales, ranging from nanoseconds to seconds and minutes (see Fig. 1).

B) Characteristic timeline of motions occurring in proteins and different suitable NMR experiments assessing these different types of
protein dynamics
The Tudor domain
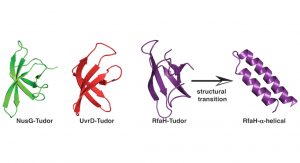
Using these powerful approaches, we recently set out to understand the inherent dynamics of a small classical protein fold, which can be found in all organisms throughout the tree of life, the Tudor domain. A Tudor domain is about ~60 amino acids in size and forms a characteristic barrel-shaped ß-strand fold (see Fig. 2). This domain mediates a variety of protein:protein interactions, but differ in their repertoire of interaction partners. In a recent study addressing the dynamics of bacterial Tudor domains, we compared them based on their functional properties; either having a small set of interaction partners (NusG)1, multiple interaction partners (UvrD)2, or metaphoric domain that can change its structure from the classical Tudor domain fold to two helices forming an inhibited state (RfaH)3 (see Fig. 2).
Our study revealed a fine-tuning of the inherent dynamical properties which could be directly coupled to the functional repertoire of the domain.4 Whereas the optimised NusG-Tudor with a defined set of interaction partners revealed the most stable fold of the proteins under study, increasing the functional repertoire for the UvrD-CTD has already shown more flexibility on the slow timescale, i.e. protein loop arrangements on the micro- to millisecond timescale, within the protein domain clearly indicating the need for local adaptations to enhance the binding promiscuity. Our compare-and-contrast study has enabled us to construct a simplified scheme correlating protein dynamics directly to functional repertoire (see Fig. 3).
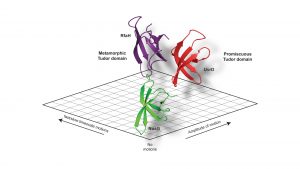
Shape-shifting properties
The most interesting finding was for the metamorphic Tudor of RfaH which, in addition to the flexibility in its loop regions, showed throughout the whole domain a large amplitude of motions on the nanosecond timescale indicative of a semi-stable protein fold.
As the understanding of the underlying driving forces of the structural switches of metamorphic proteins remain largely elusive,5 we believe that with our approaches we will be able to elucidate the functional basis of how the remarkable shape-shifting properties of this growing set of proteins can be addressed.
References
- Mooney, R.A., Schweimer, K., Rösch, P., Gottesman, M. & Landick, R. ‘Two structurally independent domains of E. coli NusG create regulatory plasticity via distinct interactions with RNA polymerase and regulators’. J. Mol. Biol. 391, 341–358 (2009)
- Kawale, A.A. & Burmann, B.M. ‘UvrD helicase-RNA polymerase interactions are governed by UvrD’s carboxy-terminal Tudor domain’. Commun Biol 3, 607 (2020)
- Burmann, B.M. et al. ‘An α-helix to β-barrel domain switch transforms the transcription factor RfaH into a translation factor’. Cell 150, 291–303 (2012)
- Kawale, A.A. & Burmann, B.M. ‘Inherent backbone dynamics fine-tune the functional plasticity of Tudor domains’. Structure (2021)
- Dishman, A.F. et al. ‘Evolution of fold switching in a metamorphic protein’. Science 371, 86–90 (2021)
Please note, this article will also appear in the seventh edition of our quarterly publication.