
The Innovation Platform spoke to Professor Sara Brucker, Executive Medical Director of the Department of Women’s Health at the University Hospital and Faculty of Medicine Tübingen, about the complexity of Mayer-Rokitansky-Küster-Hauser syndrome and how her recently-developed technique for creating a neovagina is having an positive impact on the lives of her patients.
A condition which causes the vagina and uterus to be underdeveloped or absent, although external genitalia are normal, Mayer-Rokitansky-Küster-Hauser is classified as a rare disease. This occurs in females and mainly affects the reproductive system, meaning that women with Mayer-Rokitansky-Küster-Hauser are unable to become pregnant and also experience problems with sexual intercourse.
Professor Sara Brucker, Executive Medical Director of the Department of Women’s Health at the University Hospital and Faculty of Medicine Tübingen, has recently developed a new method for the creation of a neovagina, which helps her patients to overcome this. She has also implemented the method for uterus transplantation in Germany.
The Innovation Platform spoke to Brucker about her work in this area and the successes she has seen.
Can you briefly describe your work to better understand the genetics/epigenetics behind Mayer–Rokitansky–Küster–Hauser syndrome, particularly with regard to the OXTR and ESRI genes?
Mayer-Rokitansky-Küster-Hauser syndrome was first defined some 150 years ago by those researchers who gave their name to it – Mayer, Rokitansky, Küster, and Hauser. They were investigating anatomy by studying cadavers and came to understand that there were some women who had no uterus.
Since then, the focus moved to embryology, and we now know that in the early stages (between six and eight weeks) of gestation, the internal genitals start to develop. Initially, this is independent of the sex of the child, but later on the chromosomes go on to determine the sex and, if the genetics is normal and the sex is to be female, the foetus would begin to develop a uterus. This is done via the two Mullerian ducts, which fuse together to form a single uterus in humans.
But in female foetus with Mayer-Rokitansky-Küster-Hauser we know that the fusion and subsequent development of the Mullerian ducts does not happen; they remain in their rudimentary form. Additionally, 30-40% of Mayer-Rokitansky-Küster-Hauser patients have other associated malformations, like a solitary kidney, dislocation of a single kidney (in the pelvis), or skeletal malformations like fusion defects in the spine etc. Vaginal and uterine aplasia in combination with associated malformations is classified as a Mayer-Rokitansky-Küster-Hauser syndrome type II.
If the fusion of the Mullerian ducts has begun but has stopped at a different, later stage, women could have one rudimentary uterus on one side, and a more developed one on the other; or, they could have two uteri, or perhaps one uterus that is still divided in its inner layer. So, Mayer-Rokitansky-Küster-Hauser syndrome could also be defined as the most early and therefore ‘severe’ Mullerian duct fusion or development defect. What we do know for sure is that the development of a normal uterus, cervix, and the upper two thirds of the vagina starts in the first six to eight weeks of gestation but can stop both at this early age or later on, while in ‘normal’ women, the development continues as it should.
This therefore raises the question of what causes the process to stop (regardless of the point at which it stops) and we found that, genetically, women with Mayer-Rokitansky-Küster-Hauser are totally normal – they have 46 XX chromosomes and they have normal ovaries, which means that, at around 12-14 years old, they experience puberty, with breast and pubic hair development along with the emotional development that goes with that. However, the development of their uterus stopped while they were still a foetus, and it will never develop even if their natural hormones work as they should or even if they are given hormone therapy. So, these women have a so-called ‘aplasia’ of the uterus and of the vagina, although having normal ovaries and therefore developing a normal breast (thelarche) and pubic hair (pubarche). Only the ‘menarche’, the first menstrual bleeding, cannot not occur due to the missing uterus.
That being said, we have begun to further investigate the genetics behind the syndrome. Here, we have studied five monozygotic twins where one of them has Mayer-Rokitansky-Küster-Hauser but their sister has a totally normal uterus. This demonstrates just how difficult this condition is to understand, genetically speaking. Indeed, we have also found some cases where there are multiple sisters, two of which have the syndrome, while the others are totally normal.
Because of this genetic complexity, we have been working for the last 15 years to try and find the cause of this malformation. But it has proven to be an extremely difficult task.
Nevertheless, we have gone on to establish the biggest centre in the world here in Germany dedicated to answering this question, and this has the most Mayer-Rokitansky-Küster-Hauser patients ever to attend one centre (although another of a similar size and scope is also being developed now in China). We have performed surgery on over 450 patients despite the fact that Mayer-Rokitansky-Küster-Hauser is classified as a rare disease and only occurs in just one out of 5,000 female livebirths.
The patients who come to us tend to do so during puberty because they know that there is something wrong. Perhaps they have not yet begun to have their period (because, unknown to them, they have no uterus), and we then have the very difficult job of telling them that they have no uterus and no vagina and, as such, they will never have normal sexual intercourse. Neither could they give birth to a child. They may then feel that they will never have a real relationship and will never have a boyfriend. This is devastating for them, but I am now extremely happy to be able to tell them that I can in fact create a neovagina via a minimally-invasive route, a laparoscopy. This vagina will be created from their own tissue, as the procedure involves stretching the vaginal dimple – which is typically only 0.5-2 cm – over the course of a few days to a length of 10cm. This will then create a vagina which begins to generate its own skin and which will also produce its own lubricants for sexual intercourse and so on (and, which is important for many of the patients, no sexual partner would ever be able to tell the difference between the neovagina and a natural one). This, of course, makes the patients happy, and they continue to progress through puberty in a better state of mind than when they had first been diagnosed. Because of the emotional distress that the diagnosis can often cause, we work closely with the psychosomatic department to ensure that we are also taking the patients’ mental health into account in all that we do.
Once we have informed the patient of the possibility of a neovagina and the benefits that this can have, it is great to see them begin to be themselves once more. However, in most cases the patients often come back to us a second time, again depressed, when they are 25 or 30 years old. This is because their female friends and family of the same age are now beginning to have children, and they are disappointed that they will not be able to do so too (something which is exacerbated by the fact that surrogacy is not allowed in Germany, meaning that adoption is the only solution, but the adoption of a new-born is almost impossible).
It was for this reason that we then began to look at uterine transplantation. We have completed four of these procedures now, two of which have resulted in patients having their own babies (the children are now around a year old) – the other two are currently in the process of becoming pregnant.
We believe that it is possible to successfully complete a uterine transplantation in patients with Mayer-Rokitansky-Küster-Hauser because the syndrome is not purely based on genetics; if it were, any female babies with a mother with Mayer-Rokitansky-Küster-Hauser would also have the disease, but they do not. Indeed, there have been some 60 patients with the syndrome in the USA who have had surrogate children, and yet none of them have inherited the disease.
When we begin the neovagina surgery, we explore the various relevant elements of the patient, and if we find that they have Mullerian ducts which are 2-3cm in length and complain about cyclic lower abdominal pain then we remove them so that we can explore the tissue further in an effort to better understand what is preventing them from developing properly, and if this is something that happens locally. This is important because all the other groups around the world only collect blood samples from their patients. If the underlying factor for causing the disease is indeed found locally in the tissue itself, whole genome sequencing solely of blood might not lead to the answer. Indeed, removing the Mullerian ducts means that we are able, on the one hand, to look at the RNA transcriptome, and on the other we can conduct whole genome sequencing of the tissue and are able to compare this to the patients’ blood as well as their family members, too. This gives us the unique opportunity to find out whether the disease is caused by something happening locally via, perhaps, a disturbed growth signal, or on a more global scale. I am very happy and excited that these studies were made possible through funding that I have been awarded from the German research foundation (DFG).
Apart from sequencing, we have also stimulated the tissue we have removed, and this enabled us to see that it does not proliferate and does not normally change to the secretory phase, which is what occurs in a normal endometrium. We therefore thought that there must be a disturbance in hormone receptors like the oxytocin receptor (OXTR) or the oestrogen receptor I (ESRI).
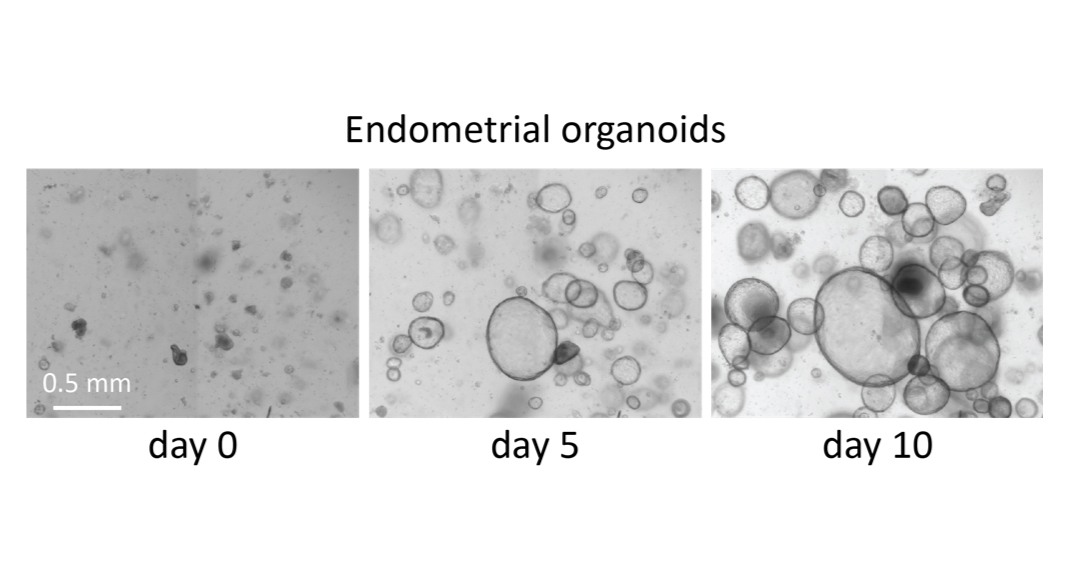
Building on this, we have continued to conduct experiments, such as those concerning the RNA transcriptome, and we hope that in the coming months we will be able to analyse the data from the whole genome sequencing that has been carried out. We were also able to establish an endometrial organoid model, which is proving to be very promising. This will give us better insights on the function of the rudiments.
You have developed an innovative new method for neovagina creation. Can you describe this? What were the main hurdles you needed to overcome, and what results have you seen?
The core principle of the method of neovagina creation was developed by a colleague in Italy, Dr Vecchietti, who was able to put tension on the vaginal dimple in order to create the neovagina. However, Vecchietti did this as an open surgery. The procedure was also very complicated. As such, we wanted to develop an alternative, minimally invasive surgery, and so we tried laparoscopy.
Perhaps the first hurdle was to undertake it with the tools that we then had at our disposal. The previously-developed procedure involved putting a dilator onto the vaginal dimple and then attaching two threads. These threads are then brought into the abdomen before being pulled behind the peritoneum and then back outside of the abdomen. A tension device is then used to add pressure on and stretch the vaginal dimple. However, the technique was not always sufficient, and we found that the threads ruptured and, in some cases, that the dilators entered the bladder.
As we wanted to develop a minimally invasive procedure, I approached one of the many medical technology companies which are based here in the south-west of Germany, and we went on to build the necessary instruments together. From that, I developed a new traction device as well as a slightly different method which meant that we can achieve a longer vagina and a better axis of the neovagina which does not go directly towards the bladder. This was significant because it reduced the risk of injuries to the bladder or rectum. After five days of tension, we found that patients then had a neovagina that measured around 10cm.
We then provide the patients with a ‘dummy’, which they are instructed to use at home with an estrogen cream for around six months. This helps the normal vaginal epithelium to develop. When we took a biopsy of the neovagina, not even the pathologist was able to tell the difference between this and a normal vagina.
Uterus transplantation can utilise organs from living or deceased donors. What are the advantages of living donation?
We are currently conducting uterine transplantation using living donors only. And while this does involve an element of risk for the donor, in every organ transplant the time for which the organ is no longer perfused with blood is of the essence, and we can ensure that this is much less when using living donors. This is not least because most deceased donors are multi organ donors and as the uterus will not be removed at first in a life-saving operation, it tends to be harvested last, which could be up to 10 hours later, therefore could dramatically reduce its potential success.
Furthermore, when using a living donor it is possible to assess both the organ and the donor in detail – we will only see the therapy as being a success if it results in a healthy baby, and not just because the uterus functions normally in terms of monthly menstrual bleeding. This includes knowing whether the donor has even been pregnant and, if so, whether there were any complications – an ectopic pregnancy, miscarriage etc., or problems during the pregnancy like hypertension, diabetes etc. We also need to know if there are myoma or fibroids, for example, and if there is an HPV infection or had been before, because the recipient of the uterus will be administered with immunosuppression and so they are at a considerably elevated risk of developing cancer from such an infection. This information can be very difficult to acquire from a deceased donor.
When we began undertaking these surgeries, no successes had been reported when using organs from deceased donors (although, since then, this has changed, with three procedures thus far going on to result in the birth of a child).
What successes have you seen since beginning the first human LD-UTx programme in Germany? What challenges have you encountered?
We have screened over 250 patients for a possible uterus transplantation, and in the end, there were only eight who really matched the criteria for the whole programme, which is mainly because of the health of the donor.
We are very happy with the results we have achieved thus far – we have completed four transplantations, and all four have a fully-functioning uterus with regular menstrual bleeding. Two of the patients have already given birth to a healthy child. Those parents would also like to have a second child, after which we will remove the uterus, meaning that they will also be able to stop taking the immunosuppression medication, which is important as it will negate the side effects that can occur from this after long-term use.
For the remaining two patients who are ready for a transplant, the COVID-19 pandemic has meant that we have had to pause our work as we are not allowed to continue IVF procedures, including performing the embryo transfer, at this time. There is a fifth patient who is also waiting to start IVF and she is waiting for a uterine transplantation. But, in a normal situation this would have started at the beginning of the year. We have also had to stop the transplantation project because of the pandemic. Hopefully, we will be able to begin these activities again towards the end of the year.
Moving forwards, perhaps once the pandemic is over and things return to normal, where will your research priorities lie? And what are your hopes for the programme in the long term?
I hope that we can move forward in the living donor programme, and it is possible that we may be able to gather enough data to be able to go on to include deceased donors, too. We will also try to make the surgery easier and reduce the time it takes for the donor, and we also plan to re-evaluate which types of immunosuppression medication we will be administering.
Furthermore, we have launched a worldwide registry for uterine transplantation which will contain data on all patients as well as any children that are born and the information from follow-ups. We hope that this will provide us with a better insight into what is the best in terms of surgical technique, immunosuppressants, how long we should prolong the pregnancy, and so on.
In addition to the clinical element, we will also advance our fundamental research in the field of Mayer-Rokitansky-Küster-Hauser. As previously mentioned, we are eager to see the analysis of the genome data especially in combination with our transcriptome data. Our plan is to then move forward with possible candidate genes and interesting signalling pathways in endometrial organoids to better understand their impact on development and growth.
My belief is that this holistic research approach will lead to a better understanding of the underlying mechanism of Mayer-Rokitansky-Küster-Hauser syndrome, if not even embryology itself, and might one day lead to the prevention of this developmental defect. In the meantime, we will continue to provide our patients with the best possible care, be it the creation of a neovagina or uterus transplantation, in order for them to have the least amount of discomfort with this disease as possible.
Professor Sara Brucker
Executive Medical Director
Department of Women’s Health
University Hospital and Faculty of Medicine Tübingen
+49 7071 29 80791
sara.brucker@med.uni-tuebingen.de
www.medizin.uni-tuebingen.de/de/dasklinikum/einrichtungen/kliniken/frauenklinik/forschungsinstitut
Please note, this article will also appear in the third edition of our new quarterly publication.






