
Dr Carlos Rogerio de Figueiredo and Professor Maija Hollmén at the University of Turku are exploiting uveal melanoma as a model system, to understand the fundamentals of immunotherapy resistance.
Immunotherapies using immune checkpoint inhibitors (ICI) have dramatically improved cancer survival in the last decade. Since the pre-clinical evidence of blocking the cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4), programmed cell death-1 (PD-1) or its ligand (PD-L1) and further successful clinical trials have proven the efficacy of ICI in inducing durable responses in some patients with advanced stage cancers such as metastatic melanoma. Essentially, ICI restores and enhances the functions of effector immune cells (e.g. cytotoxic T lymphocytes), which clear tumours mainly by inducing cancer cell death. ICI targets the immune checkpoint regulators (ICRs), mostly expressed on T lymphocytes, leading to unrestricted activation of these cells and anti-tumour functions. Therapeutic strategies using ICIs, however, are more likely to work if cytotoxic T cells are present within the tumour microenvironment (TME). This process is also reliant on the previous tumour specific clonal expansion and activation of T cells in the lymphoid organs during their interplay with antigen presenting cells (APCs), including dendritic cells (DCs) and macrophages.
Unfortunately, the majority (up to 80%) of cancer patients are refractory to ICI, or relapse these treatments by developing resistance mechanisms, often associated with an immunosuppressive TME. The tumour TME is sustained by the positive influx of tumour promoting immune cells, in which T regulatory cells (Tregs), myeloid derived suppressor cells (MDSCs), and tumour-associated macrophages (TAMs) are frequently present. Importantly, TAMs extensively populate the TME sustaining local immunosuppression and thus, immunotherapy resistance. Moreover, several studies suggest that TAMs are one of the key targets to improve the efficacy of ICI, as these cells can suppress the functions of CD8+ T and NK cells.
Uveal melanoma
In striking contrast to cutaneous melanoma, despite developing from cells of similar lineage, metastatic uveal melanoma (mUM) is almost universally refractory to ICIs. Primary uveal melanoma (pUM) is the most common intraocular cancer in adults, accounting for 5% of all melanomas. This ocular tumour develops in the choroid layer from the posterior uvea (Fig. 1), and treatment options include radiotherapy and surgery, which usually achieve local tumour control. Despite this, ~50% of pUM patients will develop metastatic disease. The average survival of mUM patients is only ~12 months, and there are currently no effective treatment options for patients with mUM. A number of clinical, histological and genetic parameters of pUM is associated with the development of metastatic disease. These include age and gender of the patient, size and location of the pUM, the presence/absence of particular histological features, as well as the loss of one copy of chromosome 3 (monosomy 3) and additional copies of the long arm of chromosome 8.1
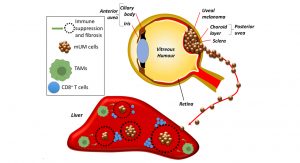
It is apparent that the tumour microenvironment is of equivalent importance to these parameters, as well as intrinsic tumour and immune cell properties in determining response to therapy and patient prognosis. Indeed, dense T-cell and macrophage infiltrates, observed in histological sections of enucleated eyes of UM patients treated with primary enucleation, are associated with a poorer outcome. Recently a TCGA study of 80 pUM demonstrated that a subset of high-metastatic risk pUM could be further stratified according to the presence of CD8+ T-cell immune infiltrates, and an altered transcriptional immune profile.1 The latter included elevated levels of HLA-I molecules, which leads to NK cell suppression, and immune checkpoint regulators (ICRs), such as PD-L1, indoleamine 2,3-dioxygenase (IDO)-1 and T-cell Ig and ITIM domain (TIGIT), which result in a predominantly immune-suppressed microenvironment. These findings suggest the potential use of ICIs in mUM, however, no benefits to patient outcome have been observed with these agents so far.
Properties of Ocular immune privilege
The majority of mUM spreads into the liver, which is an environment offering tolerance to non-self-antigens by suppressive macrophage populations leading to incomplete immune cell activation (Fig. 1). While immunotherapy resistance in mUM has been partly ascribed to a low mutational burden, responses to adoptive cell therapy using tumour infiltrating lymphocytes, and the presence of an immune infiltrate (albeit partially excluded from the interior of mUM) suggest this is not the sole reason. pUM and mUM appear to have intrinsic properties, which enable them to escape or be camouflaged from the immune surveillance systems within the body. These intrinsic properties could originate from the microenvironment of the eye, which is an immune privileged site. Such sites including also the placenta, brain, and testicles can tolerate the introduction of antigens without eliciting an inflammatory immune response. This protects vital eye structures from any uncontrolled inflammation in order to protect vision.
Several evidences have suggested that the putative immune privilege of the eye somehow educates or gives the capacity of UM cells to also tolerise their metastatic microenvironment differently from tumour cells spreading from non-immune privileged sites. Therefore, investigating the behaviour of UM cells in their primary and metastatic microenvironment gives us the opportunity to define factors contributing to immune escape and refractoriness to ICI therapy.
Importance of TAMs in the pathology of UM
The mechanism underlying the ‘immune camouflage’ behaviour of UM is poorly understood. It is known that CD4+ and CD8+ T cell infiltrates are present in mUM, but they are usually peritumoural and appear to be unable to penetrate into the tumour mass interiors and to provide antitumour effects. There is frequently a wall of sclerotic tissue surrounding the mUM, isolating them from the adjacent liver parenchyma and encroaching inflammation. Research has shown that pUM tumours that include high levels of infiltrated TAMs are associated with poor survival. These TAMs usually express CD163, a macrophage marker for the anti-inflammatory phenotype, which is associated with pro-tumourigenic functions, metastasis and immune suppression in many cancers. Since liver metastases from UM can remain dormant for several years before they become clinically detectable, TAMs supposedly contribute to their growth in the hepatic microenvironment, justifying the development of adjuvant strategies targeting TAMs in the liver and enabling modification of the tumour microenvironment and its immunologic properties.
In the tumour microenvironment, TAMs can secrete soluble factors in response to cancer cell stimuli, which contributes to the metastatic process and immunosuppression through Treg and myeloid derived suppressor cell (MDSC) recruitment. Among these factors, TAMs may also directly suppress effector T cell responses by co-expressing myeloid checkpoint regulators (ICR), including Clever-1. Clever-1, also known as Stabilin-1, is a multifunctional molecule conferring scavenging functions on a subset of immunosuppressive macrophages.2 Our group initially showed that the progression of cutaneous melanoma growth and metastasis is regulated by Clever-1. Therapeutic treatment of mice with an anti-Clever-1 neutralising antibody inhibited tumour progression of melanoma, lymphoma, lung, breast cancer and colon carcinoma in vivo.3-5 Particularly, the treatment of a triple-negative breast cancer and colon carcinoma model with anti-Clever-1 antibody not only increased the levels of tumour infiltrating CD8+ T cells, but also improved the survival responses to anti-PD1 blockade in these tumours, suggesting that Clever-1 expression in the stroma is associated with intrinsic mechanisms of cancer immunosuppression and immunotherapy resistance.5
Current and future treatments for mUM
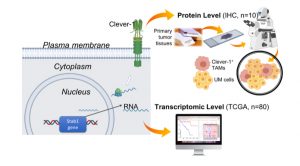
We recently evaluated the expression levels of Clever-1 in pUM tumours, either in the RNA level of the TCGA UM data set (Stab1 gene), and in a smaller cohort of pUM tissues at the protein level by immunohistochemistry (IHC) (Fig. 2).
Importantly, we observed that high Clever-1/Stab1 expression is significantly associated with the poorest prognosis, and thus metastasis development and low patient survival (Fig. 3). Therefore, patients that express high levels of Clever-1 are likely to be refractory to immunotherapies using ICI. Although immunotherapy has been widely used with established approved agents, it has been disappointing to date in mUM. The response rates in CM to single-agent anti-CTLA4 (ipilimumab) and anti-PD-1 (nivolumab or pembrolizumab) are in terms of 10–20 and 40%, respectively, but in mUM single-agent therapy has been reported to reach response rates of 5% or less.
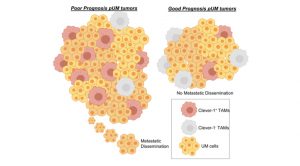
However, a recent clinical approach with adoptive transfer of tumour infiltrating lymphocytes (TILs) revealed that tumour specific T cells are present in mUM and that these cells may be stimulated to engender clinically meaningful responses. In an indirect way, the tumour microenvironment could also be targeted for specific pathways in order to unleash the anti-tumour effects of TILs.
An alternative approach to ICI, is the use of a novel bispecific molecule, IMCgp100 (Tebentafusp), which is under clinical development (Immunocore, OXF, GB). One arm of the IMCgp100 molecule is an enhanced T-cell receptor targeting the overexpressed gp100 molecule, which is bound to the HLA A201 in mUM, while the other arm of the molecule is an anti-CD3 scFv. This construction enables IMCgp100 to redirect T-cells into mUM tumours, leading ultimately to T-cell activation and cytotoxicity against the tumour. Two Phase I trials have reported initial results in UM (NCT01211262 and NCT2570308), suggesting disease stabilisation in a significant fraction of patients and a 1-year overall survival of 73% in both studies, which is significantly higher than expected for this population of patients. However, this approach is not invulnerable to the myeloid immune resistant mechanisms raised in the TME, such as those triggered by Clever-1+ TAMs.
The pre-clinical studies with Clever-1 have led to the clinical development of a humanised anti-Clever-1 antibody (FP-1305). The MATINS (Macrophage Antibody to FP-1305 To INhibit immune Suppression) study (NCT03733990) is a first-in-human open label Phase I/II adaptive clinical trial (sponsored by Faron Pharmaceuticals) in selected metastatic or inoperable solid tumours to investigate the safety and efficacy of FP-1305. Because of our prognostic findings of Clever-1 in uveal melanoma outcome, it was selected as a patient cohort together with cutaneous melanoma, hepatobiliary, pancreatic, ovarian, ER+ breast, colorectal, gastric, gallbladder cancer and cholangiocarcinoma to investigate the efficacy of FP-1305 in part II of the phase I trial.
Metastatic UM constitutes one of the most challenging tumour types and is relatively homogenous with few differences between patients in terms of mutational landscape and a well-defined clinical behaviour. As such, it is an ideal model to investigate tumour immune evasion and resistance to ICI therapies. Insights from our current studies of Clever-1 in UM enable the clinical development of a macrophage targeted immune therapy for mUM, but will also be applicable to our understanding of the fundamental mechanisms of immune escape in cancer.
References
1 A. G. Robertson et al., Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 32, 204-220.e215 (2017)
2 J. Kzhyshkowska, A. Gratchev, S. Goerdt, Stabilin-1, a homeostatic scavenger receptor with multiple functions. J Cell Mol Med 10, 635-649 (2006)
3 K. Schönhaar et al., Expression of stabilin-1 in M2 macrophages in human granulomatous disease and melanocytic lesions. Int J Clin Exp Pathol 7, 1625-1634 (2014)
4 M. Karikoski et al., Clever-1/stabilin-1 controls cancer growth and metastasis. Clin Cancer Res 20, 6452-6464 (2014)
5 M. Viitala et al., Immunotherapeutic Blockade of
Macrophage Clever-1 Reactivates the CD8. Clin Cancer Res 25, 3289-3303 (2019)
Maija Hollmén, PhD
Group Leader
MediCity Research Laboratory,
Institute of Biomedicine,
University of Turku
+358 50 514 2893
maija.hollmen@utu.fi
Tweet @maijahollmen @HollmenMaija




