
Professors Andres M Gomez and Marshall D Stern, from the University of Minnesota, and Isaac Salfer from South Dakota State University, outline how a better understanding of the relationship between the rumen microbiome and nutrient digestion in dairy cows is being developed.
Ruminant animals have been an important component of human agriculture for over 10,000 years. Ruminant species including cattle, sheep, and goats continue to be an important source of food and fibre throughout the world today (Zeder & Hesse, 2000). One of the primary reasons why ruminants have been indispensable in human society is because they have the unique ability to convert fibrous materials such as grasses and forages that are indigestible by any mammal into high-quality human-consumable protein. This ability is generated by the diverse array of micro-organisms including bacteria, archaea, protozoa, and fungi that exist within the ruminant foregut. These microbes possess enzymes that allow them to degrade and capture energy from complex carbohydrates including cellulose, hemicellulose, and pectin. The ruminant animal uses volatile fatty acids (VFA), which are produced as a by-product of fibre fermentation, to meet their own energy requirements. These VFA include acetate, propionate, and butyrate, which represent the primary source of energy for ruminant metabolism. Rumen microbes can also utilise non-protein nitrogen (NPN) sources such as urea, nitrates, or nucleic acids to synthesise their own amino acids for growth and milk production. As rumen microbes pass out of the abomasum referred to as the ‘true stomach’, microbial cells are digested and absorbed from the small intestine as amino acids. These amino acids help the animal meet over half of their requirements for maintenance, growth, reproduction, and lactation.
Developing an improved understanding of fermentation within the rumen, the compartment of the foregut where the majority of nutrient digestion occurs, can provide tremendous opportunities to improve the efficiency of ruminant livestock production and reduce the environmental impact of ruminants. Although researchers have been studying rumen metabolism for over a century, many opportunities remain to improve rumen efficiency and the efficiency of ruminant livestock production. Improving the capture of nutrients by the rumen can lead to the reduction of greenhouse gasses by ruminants, which will lower the contribution of ruminants to global climate change. The development of new analytical techniques that allow for rapid quantification of the entire rumen microbial function and a wide complement of rumen metabolites allows us to understand the rumen in ways that were never possible before. These technologies, typically referred to as ‘omics’ techniques, coupled with mechanistic in vitro studies, create opportunities to develop a fundamental understanding of the biochemical pathways in the rumen which will lead to development of feeding strategies to reduce inefficient rumen fermentation.
The rumen microbiome: a highly efficient system to digest feed nutrients
The rumen microbiome is composed of a highly diverse group of micro-organisms that includes a large proportion of bacteria, followed by ciliated protozoa, anaerobic fungi, bacteriophages (viruses), and methanogenic archaea which are the only micro-organisms capable of producing methane. This anaerobic micro-ecosystem has an abundance of up to 100 billion microbial cells per gram of ruminal content in an 80 litre rumen chamber, representing from 1/7th to 1/10th of the total weight of the ruminant animal. Rumen micro-organisms form a complex network of metabolic pathways that efficiently convert ingested feed into various compounds. These compounds are used by the microbes to survive within the rumen chamber and simultaneously provide the animal with indispensable nutrients for growth and development. This mutualistic relationship and the high efficiency of the rumen microbiome to digest feed is what defines the value of ruminants as critical food sources to feed a growing human population.
Rumen microbes enable the digestion of complex fibrous materials (cellulose, hemicellulose, and pectin) in grasses and forages by producing fibrolytic, glycolytic, lipolytic, and proteolytic enzymes. These enzymes facilitate VFA as a main product of feed metabolism and fermentation. Lactate, hydrogen (H2) and carbon dioxide (CO2) are also produced from rumen fermentation. These metabolites feed back into microbial metabolism and/or are released by the animal in the form of methane (generated by rumen methanogenic archaea) to ensure the energetic efficiency of the VFA generation process. The potential of rumen microbial enzymes to metabolise the most recalcitrant fibres and release energy rich compounds is such that the biofuel industry often mines the rumen microbiome for novel enzymes that can help make use of industrial and agricultural byproducts. Microbially-produced VFA mainly acetate, propionate, and butyrate, can be produced in ratios approximately 75:15:10 to 40:40:20, respectively. The ratio of VFA produced in the rumen depends largely on diet, with high-fibre diets resulting in greater acetate concentrations, and low-fibre diets resulting in higher propionate concentrations. Volatile fatty acids contribute up to 70% of the total energetic requirements of ruminants once absorbed through the rumen epithelial cells (Bergman, 1990). Thus, feed conversion in ruminant production systems is inextricably linked to rumen microbiome function and its feed degrading efficiency. For these reasons, the rumen microbiome is perhaps one of the most critical targets of management and feeding strategies aimed at improving the efficiency of beef and dairy production systems.
The rumen ecosystem has been studied for more than 50 years. Recently, there has been greater implementation of modern DNA sequencing techniques and other high-throughput molecular and computational approaches used to characterise diversity and function of rumen microbes, also referred as ‘omic’ techniques. Utilisation of these techniques allows linking of a myriad of rumen microbiome traits to various productive parameters.
The vast compositional and functional diversity of the rumen microbiome
Pioneering work by Drs Robert Hungate and Marvin Bryant in the 1950s paved the way for understanding the compositional and functional characteristics of the rumen micro-ecosystem. Using anaerobic in vitro culture methods, they identified and characterised major rumen bacterial groups with critical roles in feed digestion such as Prevotella, Ruminococcus, Butyrivibrio, Fibriobacter, and Selenomonas among many others. Their research elucidated how major rumen bacteria metabolise feed substrates and their contributions to VFA generation. However, a major limitation of culture-dependent approaches is that not all rumen bacteria can be currently grown in vitro, which misses a substantial fraction of rumen micro diversity. Thus, in the early 1990s the study of microbial ecology advanced significantly with the implementation of molecular techniques that relied on sequencing specific DNA and RNA markers of micro-organisms in any ecosystem to infer their diversity, abundance, and potential functions. This advance reached its full potential when sequencing techniques became high-throughput and when computational and data analysis methods were made widely available to the research community, allowing us to characterise the compositional and functional diversity of the rumen microbiome across hundreds and thousands of samples (animals) simultaneously and with a resolution not available based on culture-dependent methods.
High-throughput nucleic acid sequencing methods have become the gold standard in studying the rumen microbiome, with several options depending on the question in mind. For instance, sequencing can focus on a fragment or short amplicon of specific taxonomic marker genes in bacteria, archaea (the 16S ribosomal (r)RNA gene), fungi, and protozoa (18S rRNA or internal transcribed spacer- ITS1), which reveals the diversity and composition of a given micro-ecosystem. However, sequencing does not offer any information on the functions performed by those micro-organisms. By randomly surveying small fragments of other genes, a technique known as shotgun metagenomics or metatranscriptomics sequencing (in the case of messenger (m)RNA characterisation), we can obtain a view of the functions potentially performed by all the resident microbial communities, along with information of the most active rumen microbes, in the case of transcriptomic analyses. The resolution of nucleic acid-based techniques is such that their use has unveiled several bacterial groups thought to be potentially pathogenic or unimportant for rumen metabolism (members of Proteobacteria such as Acinetobacter, Succinimonas, Succinivibrio), while revealing more information on the diversity, abundance and function of protozoa, fungi, bacteriophages, and archaea.
Other high-throughput molecular techniques such as metabolomics and metaproteomics can provide a better estimate of the functions performed by rumen microbes. Metabolomics relies on mass spectrometry, liquid chromatography, or/and nuclear magnetic resonance techniques that can identify metabolites or chemical compounds produced by rumen microbes, by animals or co-metabolites derived from the metabolism of feed by both microbes and animals. Examples of the kinds of metabolites that can be identified through metabolomics are major substrates and products of the feed fermentation process such as lipids, mono and polysaccharides amino acids, VFA, lactate, methane, amines, conjugated fatty acids, branched chain fatty acids, carboxylic acids, phospholipids, cholesterol esters, antibiotics, and phenolic compounds, among many others.
Metaproteomics, also based on mass spectrometry-liquid chromatography methods, provides a more direct overview of the specific functions performed by microbes by quantifying peptides making part of the microbial enzymatic machinery. This method can also provide a measure of the most active microbes in rumen function and the enzymatic mechanisms by which they contribute to feed digestion. One drawback of these meta-omic techniques is that they highly depend on genetic and chemical reference databases for structural and functional identification, which can result in a great proportion of unidentified features based on the fact that the rumen is still a novel reservoir of microbes and chemical compounds not identified before. Although more efforts have been devoted to fully characterise the genomes of major rumen microbes, a large proportion of rumen micro-organisms remain uncharacterised due to the impossibility of growing them in vitro (~>60%). Regardless, the combined use of high-throughput nucleic acid sequencing and chemical techniques as applied today offer an unprecedented depth to understand the structural and functional complexity of the rumen micro-ecosystem. More importantly, they facilitate inferences of direct associations between specific rumen microbiome features and animal performance.
Complexity of the rumen microbial ecosystem in the context of animal performance
A myriad of studies based on meta-OMIC techniques have linked rumen microbiome and animal performance traits. These techniques have made it possible to select specific rumen microbiome features that explain between 40-60% variation in feed conversion efficiency, average daily weight gain, residual feed intake, and daily feed intake among animals (Lima et al., 2019; Auffret et al., 2020). The microbial features most likely associated with these productive parameters mainly reflect microbial functions involved in cellulose and hemicellulose degradation, maintenance of microbial growth, synthesis of vitamins (e.g. B12), and an increase in the generation of energy-rich metabolites (e.g. butyrate and propionate). More efficient animals in dairy systems have also shown lower rumen taxonomic and functional diversity (but increases in dominance of specific bacterial groups), higher abundance of total VFA produced, increases in generation of butyrate and propionate (from succinate and lactate) and lower methanogenesis (Shabat et al., 2016). Moreover, specific shifts in the abundance of major bacterial groups such as Lachnospiraceae and Prevotellaceae have been associated with the percentage of fat in milk and overall milk yield (Jami et al., 2014; Lima et al., 2015).
Other performance parameters that can effectively be associated with specific rumen microbiome traits include response to specific feed additives, ruminal disorders, and methane production. Similar to other mammalian microbiome systems, there is very strong microbiome variation amongst animals, meaning individual animals may exhibit very unique rumen microbiome profiles. This observation is in part due to an influence of cow genetics in shaping the composition and function of the rumen microbiome (Li et al., 2019). In the case of methane emissions, it has been found that variation in CH4 production is influenced by both individual host (cow) genotype, and the rumen microbiome composition. This observation indicates that reducing CH4 emissions in ruminants should include both selective breeding and strategies that directly modulate the rumen microbiome, such as the use of feed additives. Likewise, there have been reports of individualised responses of bovines to rumen transfaunation, with rumen microbial communities returning to pre-transfaunation states in an individual manner (Zhou et al., 2018). Because this inter-individual variation cannot be circumvented in actual in vivo production settings, the use of in vitro models that mimic the rumen environment provide a powerful approach for evaluating effects of different feeding interventions on rumen function, free from biases imposed by large differences in the rumen microbiome among different animals.
Dual-Flow Continuous Culture Fermenters
Several challenges exist when studying the impact of dietary interventions on rumen fermentation and rumen microbiology in live animals. Genotypic and phenotypic differences among animals can introduce variation into in vivo experiments. Furthermore, in vivo experiments are expensive, labour intensive, time consuming, and subject to error associated with use of digesta flow rate markers, microbial markers and inherent animal variation (Stern et al., 1997). Researchers must also consider the welfare of the animals during in vivo experiments, which can limit the number of animals used, or limit the type of treatments administered.
One model that can be used to study rumen fermentation and microbial ecology without the limitations that accompany in vivo experiments is the dual-flow continuous culture fermenter system (https://researchfeatures.com/2018/10/05/exploring-microbial-digestive-dynamics-ruminants-in-vitro/). This system contains microbes harvested directly from the rumen of a live animal and maintains them in a fermentation flask that receives constant inputs of feed and buffers and constantly removes fermentation products (see Fig. 1). Dual-flow continuous culture fermenters allow researchers to study the impacts of novel feeds, feed additives, and other compounds on rumen fermentation and rumen microbial ecology in a much quicker, safer, and more cost-effective manner than in vivo experiments (Stern & Gomez, 2018). Moreover, researchers can use fermenters to study the impact of treatments that would not be feasible in animals such as feeds or feed additives that have not yet been approved for animal consumption, or extreme concentrations of treatments that may cause risk to animals.
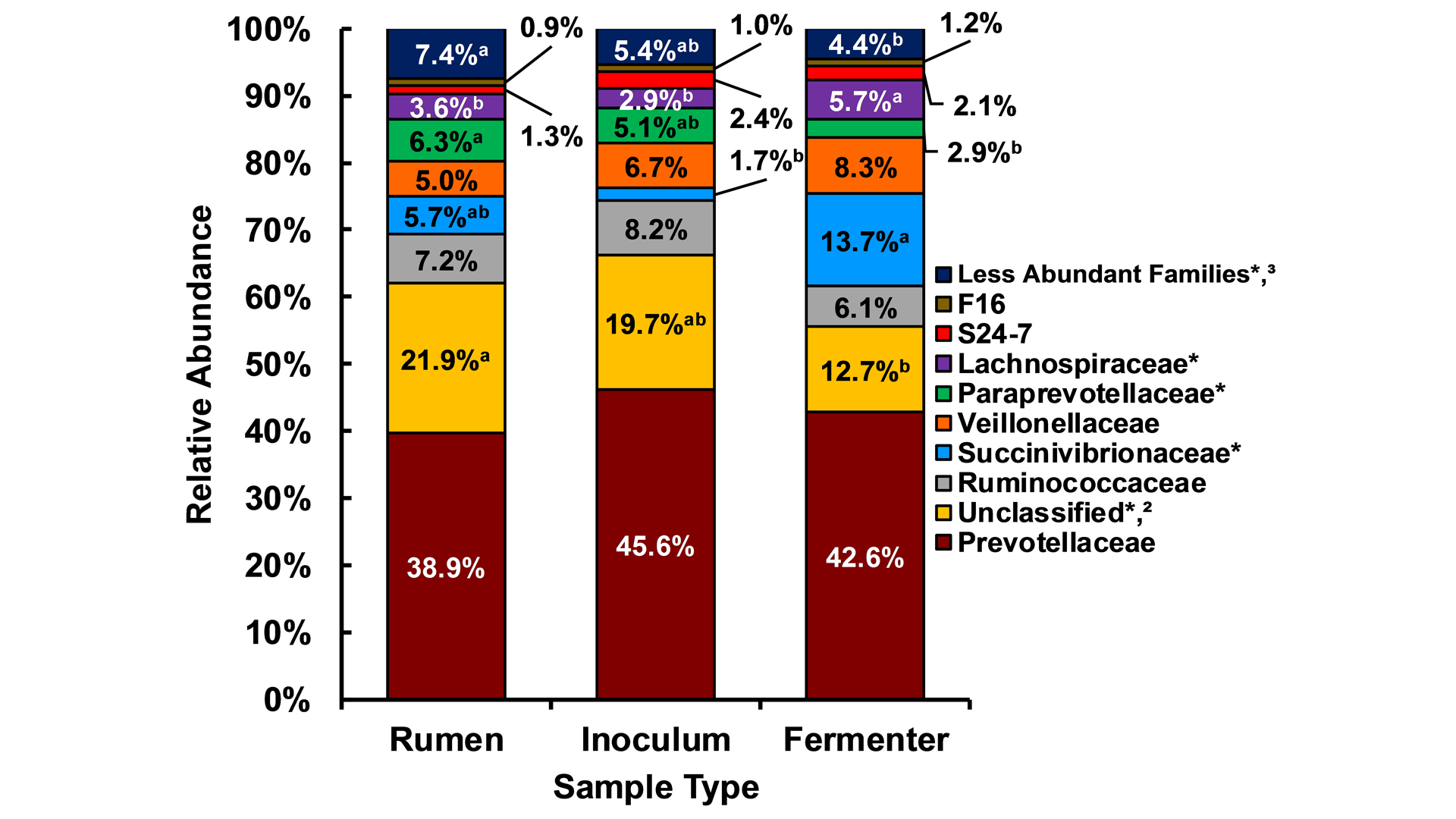
Previous research has demonstrated the efficacy of the dual-flow continuous culture fermenter system for modelling ruminant digestion, microbial fermentation, and microbial community (Hoover et al., 1976; Mansfield et al., 1995). More recently, we determined using ribosomal RNA-based sequencing of rumen bacterial and archaeal populations that many of the most highly abundant microbial families are maintained in continuous culture at concentrations similar to those found in the rumen (Salfer et al., 2018). Specifically, Prevotellacaea and Ruminococcaceae, the two most prominent microbial families with known roles in protein and fibre digestion, did not differ in abundance between natural rumens and fermenters (see Fig. 2). This experiment demonstrated that continuous culture fermenters can serve as an excellent model to complement meta-omic techniques to understand how different feeds and feed additives can modify the rumen microbiome.
Conclusion
The rumen microbiome is a critical target of strategies focused on improving the efficiency and sustainability of dairy and beef production systems. In particular, the use of meta-OMIC techniques have significant promise to understand the functional landscape of the rumen microbiome and its impact on animal physiological performance. Currently, our efforts focus on exploring modes of action of probiotics, prebiotics, enzymes, and other feed additives that can trigger changes in the rumen microbiome to reduce methanogenesis in ruminant animals, while improving feed efficiency. To accomplish this goal, we use in vitro, continuous culture fermenters, which allow for consistent, accurate measurements of microbial activity and its metabolic outputs.
References
Auffret, M. D., Stewart, R. D., Dewhurst, R. J., Duthie, C.-A., Watson, M., & Roehe, R. (2020). ‘Identification of microbial genetic capacities and potential mechanisms within the rumen microbiome explaining differences in beef cattle feed efficiency’. Frontiers in Microbiology, 11(June), 1–16. https://doi.org/10.3389/fmicb.2020.01229
Bergman, E. N. (1990). ‘Energy contributions of volatile fatty acids from the gastrointestinal tract in various species’. Physiological Reviews, 70(2), 567–590. http://www.ncbi.nlm.nih.gov/pubmed/2181501
Hoover, W. H., Knowlton, P. H., Stern, M. D., & Sniffen, C. J. (1976). ‘Effects of differential solid-liquid removal rates on fermentation parameters in continuous cultures of rumen contents’. Journal of Animal Science, 43(2), 535–542. https://doi.org/10.2527/jas1976.432535x
Jami, E., White, B. A., & Mizrahi, I. (2014). ‘Potential role of the bovine rumen microbiome in modulating milk composition and feed efficiency’. PloS One, 9(1), e85423. https://doi.org/10.1371/journal.pone.0085423
Li, F., Li, C., Chen, Y., Liu, J., Zhang, C., Irving, B., Fitzsimmons, C., Plastow, G., & Guan, L. L. (2019). ‘Host genetics influence the rumen microbiota and heritable rumen microbial features associate with feed efficiency in cattle’. Microbiome, 7(1), 1–17. https://doi.org/10.1186/s40168-019-0699-1
Lima, F. S., Oikonomou, G., Lima, S. F., Bicalho, M. L. S., Ganda, E. K., de Oliveira Filho, J. C., Lorenzo, G., Trojacanec, P., & Bicalho, R. C. (2015). ‘Prepartum and postpartum rumen fluid microbiomes: Characterization and correlation with production traits in dairy cows’. Applied and Environmental Microbiology, 81(4), 1327–1337. https://doi.org/10.1128/AEM.03138-14
Lima, J., Auffret, M. D., Stewart, R. D., Dewhurst, R. J., Duthie, C.-A., Snelling, T. J., Walker, A. W., Freeman, T. C., Watson, M., & Roehe, R. (2019). ‘Identification of rumen microbial genes involved in pathways linked to appetite, growth, and feed conversion efficiency in cattle’. Frontiers in Genetics, 10(JUL), 1–18. https://doi.org/10.3389/fgene.2019.00701
Mansfield, H. R., Endres, M. I., & Stern, M. D. (1995). ‘Comparison of microbial fermentation in the rumen of dairy cows and dual flow continuous culture’. Animal Feed Science and Technology, 55(1–2), 47–66. https://doi.org/http://dx.doi.org/10.1016/0377-8401(95)98202-8
Salfer, I. J., Staley, C., Johnson, H. E., Sadowsky, M. J., & Stern, M. D. (2018). ‘Comparisons of bacterial and archaeal communities in the rumen and a dual-flow continuous culture fermentation system using amplicon sequencing’. Journal of Animal Science, 96(3), 1059–1072. https://doi.org/10.1093/jas/skx056
Shabat, S. K. Ben, Sasson, G., Doron-Faigenboim, A., Durman, T., Yaacoby, S., Berg Miller, M. E., White, B. A., Shterzer, N., & Mizrahi, I. (2016). ‘Specific microbiome-dependent mechanisms underlie the energy harvest efficiency of ruminants’. The ISME Journal, 10(12), 2958–2972. https://doi.org/10.1038/ismej.2016.62
Stern, M. D., Bach, A., & Calsamiglia, S. (1997). ‘Alternative techniques for measuring nutrient digestion in ruminants’. Journal of Animal Science, 75(8), 2256–2276. https://doi.org/10.2527/1997.7582256x
Stern, M. D., & Gomez, A. M. (2018, October). ‘Exploring microbial digestive dynamics of ruminants in vitro Behind the Research’. Research Features, 22–25. https://researchfeatures.com/2018/10/05/exploring-microbial-digestive-dynamics-ruminants-in-vitro/
Zeder, M. A., & Hesse, B. (2000). ‘The initial domestication of goats (Capra hircus) in the Zagros mountains 10,000 years ago’. Science, 287(5461), 2254–2257. https://doi.org/10.1126/science.287.5461.2254
Zhou, M., Peng, Y. J., Chen, Y., Klinger, C. M., Oba, M., Liu, J. X., & Guan, L. L. (2018). ‘Assessment of microbiome changes after rumen transfaunation: implications on improving feed efficiency in beef cattle’. Microbiome, 6(1), 62. https://doi.org/10.1186/s40168-018-0447-y
Andres M Gomez
Assistant Professor of Microbiomics
University of Minnesota
+1 612 624 9744
gomeza@umn.edu
www.ansci.umn.edu
Please note, this article will also appear in the fourth edition of our new quarterly publication.






